COVID-19 Research FAQs Archive
Phase 4:
The following FAQs provide Phase 4 specific information that is supplemental to the Human Subjects Research Phases Table.
Please read these FAQs before submitting an Individual PI Plan to resume or initiate Phase 4 research.
What general considerations apply for human subjects research which may qualify to resume during Phase 4?
Researchers must meet the requirements of the VPR Resumption of Research Phased Plan (training & individualized plan submission for Phases 2-4) before conducting any human subjects research. Both the researcher training completion and the individualized plan for research workspace/personnel are automatically routed by the VPR’s office to the respective department chair for evaluation. Each potential point of transmission risk should be carefully considered and detailed in the investigator’s plan, including clear descriptions of the ways that risk will be mitigated/controlled. Submit plans to resume or conduct research qualifying for Phase 4 at https://uky.az1.qualtrics.com/jfe/form/SV_0V3V83mWOAzdDzn.
Generally, conditions for research conducted in the community, inside the home of an individual human participant, or at an external facility, are outside of the investigator’s control. In-person research in which transmission risks cannot be adequately controlled by the research teams, requires Pre-Approval by the Office of the Vice President for Research. Plans should address safeguards within the researcher’s capability, such as maximization of PPE and rigorous screening protocols.
The same guiding principles of social distance/physical barriers, appropriate personal protective equipment (PPE), and performance of remote human subjects research activities, whenever possible, remain as first-choice options for all research eligible in Phase 4. In addition, the VPR Phased Resumption Plan includes Key Points for Minimizing COVID-19 exposure and transmission at all phases (see Health and Safety Measures for specific workspace guidance).
What specific restrictions or requirements are there for Phase 4 research?
Research conducted during Phase 4 must continue efforts to mitigate risk of transmission:
- Research with individuals who are “at risk” (e.g., elderly, pre-existing conditions) in which participation does not offer direct benefit to the individual should employ maximum safeguards to protect “at risk” volunteers.
- In-person interaction involving physical contact, should continue to employ safeguards such as minimal time exposure with a single participant, minimal staff present and applicable personal protective equipment (PPE) (e.g., N95 masks, gowns, gloves, eye protection or face shields or barriers). Brief interactions are less likely to result in transmission of virus. Thus, decisions should be made consistent with current CDC recommendations (https://www.cdc.gov/coronavirus/2019-ncov/php/public-health-recommendations.html),
- Where possible, research with in-person group meetings of 10 or more should be conducted outdoors or in facilities allowing a minimum 6-foot social distancing or physical barriers and adequate air exchange.
- Regarding procedures involving aerosolized saliva, the American Thoracic Society recommends that pulmonary function testing be limited only to cases that are essential for immediate treatment decisions, and that measures to protect both the staff and individuals being tested should be put in place. Research procedures that result in aerosolizing saliva include, for example, sputum induction, pulmonary function tests in nonclinical space, sputum culture, pulmonary exercise tests, vigorous exercise resulting in increased and/or forceful respiration, and singing. The following protective measures are recommended: PPE that limits aerosolized droplet acquisition for staff (mask, eye protection, shield, gown, gloves), and enhanced sanitizing of the testing space. Other in-door research procedures or activities that result in aerosolizing saliva (e.g., vigorous exercise, singing) are advised to implement similar protective measures.
In transitioning to Phase 4, are there specific human participants research populations that warrant additional safeguards or support?
During human interaction, researchers should apply universal precautions as anyone is a potential carrier of COVID-19. However, the CDC has defined populations that may need extra support, access, or precautions (e.g., homeless people, people with disabilities, refugees, etc.). Population specific guidance is available at https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/index.html.
Can undergraduates serve as research personnel in human participant research studies that take place during Phase 4?
Consistent with the VPR Plan (Section D), during Phase 4, graduate students and postdoctoral fellows continue research. Undergraduates can participate in research but[SB1] should be re-tested prior (7 days) to entering the laboratory and PI plans should be updated to include these and other researchers within their programs during phase 4, with attention to following safety guidelines. Human participants research that poses special situations where risk mitigation is complicated can be performed with special pre-approval (which requires submission of a revised PI plan describing the research personnel, space usage and approach needed to reduce risk as much as possible). In general, high school students are not allowed in research workspaces during Phase 4 unless they have been pre-approved within revised PI plans. This would include requirements for testing through UK Health Corps and receiving information on safety practices and procedures when performing research during the COVID-19 pandemic.
Phases for Resumption of Human Subjects Research
-
What is the current status of human subjects research at UK?
-
How do I determine if I can conduct in-person research?
-
Human Subjects Research Phases Table
-
Once I am permitted to resume the paused research, will there be any restrictions on locations?
-
What if my research study is included in the “allowed” category, but I am not able to comply with precautions and safety guidelines related PPE, physical distancing, and maximum occupancy in a facility?
-
When can I resume my community-based research?
-
What are some issues I should be aware of when resuming community-based research?
-
What precautions should I take if I am conducting in-person research?
-
Should I implement any special procedures in my research space to protect study personnel and research participants from exposure?
-
Do I have to call to pre-screen all participants before they come in for a scheduled research visit?
-
What screening protocol should I use when interacting with research participants?
-
Are positive COVID-19 test results reportable to the state of Kentucky?
-
After research reopens if a subject or research team member is discovered to have tested COVID positive or presumptive positive with 2 weeks of an in-person research visit, would we promptly report to the IRB as an Unanticipated Problem involving Risks to Subjects or Others (UPIRSO)?
-
What do I need to know to submit/conduct COVID-19 research?
-
What is the PREP Act and what should subjects in COVID-19 interventional trials be told in the informed consent about liability limits?
-
Are there other resources I can view for further guidance?
-
What if my research involves a public health authority or public health surveillance?
-
What public health surveillance activities are deemed not to be human subject research under the revised Common Rule?
What is the current status of human subjects research at UK?
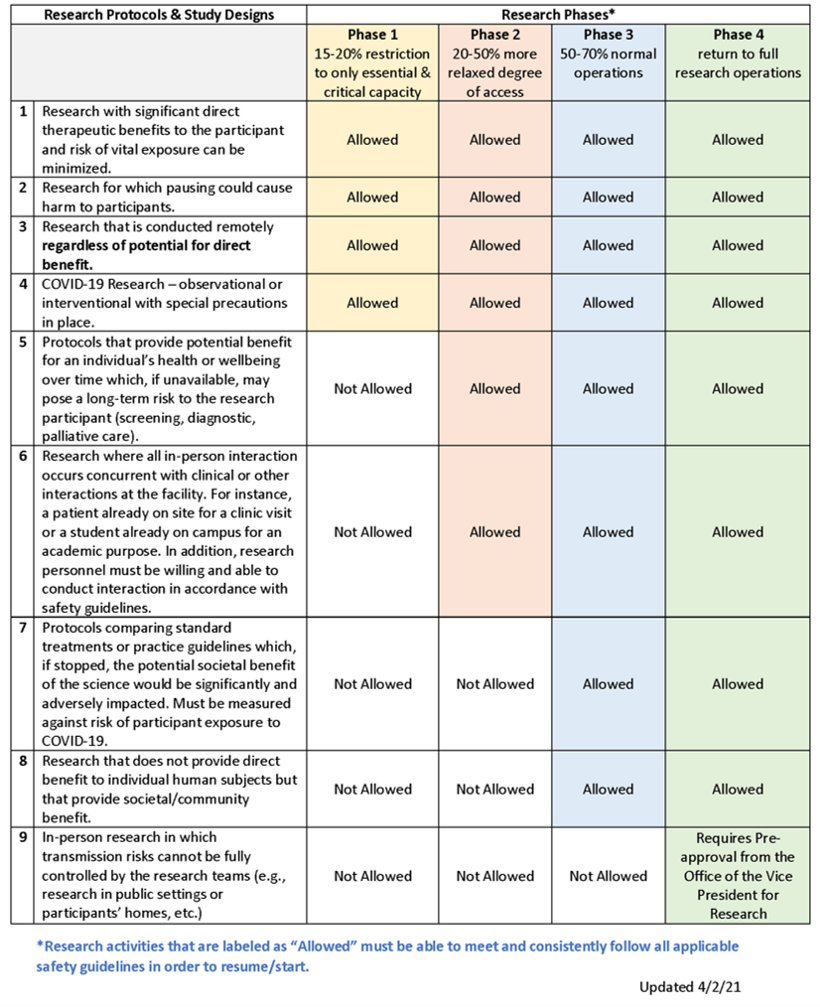
Please refer to the VPR’s University of Kentucky Resumption of Research Phased Plan for guidance on the current phase and the ORI Human Subjects Research Phases Table to determine if your human research is allowed to resume in the current phase after meeting all requirements outlined below.
How do I determine if I can conduct in-person research?
This determination is a combination of:
- Meeting the requirements outlined in the VPR Resumption of Research Phased Plan (i.e., waiver for Phase 1, training & individualized plan submission for Phases 2-4);
- Compliance with the Human Subjects Research Phases Table; and
- Permission from your Department Chair or Center Director.
The phased approach and these guidelines provide a baseline framework from which additional variables must be considered.
- Continuing or resuming human research is contingent on active COVID-19 cases, available testing and contact tracing, and compliance with health authority guidance from national, state, local, and University authorities.
- Polices and guidance will be dynamic and may accelerate or decelerate based on shifting information and incidence data. Researchers must be prepared to halt activities on short notice if this becomes necessary based on new information.
- Resuming human research operations must be parallel to and consistent with the institutional and VPR’s plans, as well as specific facility, college, and/or unit mandates.
Investigators may choose to be more stringent or cautious than phases allow.
It is critical to keep research participants informed of study status and changes that could impact them. Respect participant wishes regarding resuming in-person interaction.
Human Subjects Research Phases Table
Once I am permitted to resume the paused research, will there be any restrictions on locations?
Please refer to the VPR Resumption of Research Phased Plan for UK locations. For other research locations/facilities, determine whether those locations/facilities have any restrictions or requirements for human subjects research. With pre-approval, human participants research that takes place in different settings such as homes, churches or other parts of the community, may resume; however, mitigating risk as much as possible will require advance coordination, contingency planning and responsiveness to evolving situations.
What if my research study is included in the “allowed” category, but I am not able to comply with precautions and safety guidelines related PPE, physical distancing, and maximum occupancy in a facility?
Research must be included in the allowable category AND meet safety guidelines to resume operations. This includes PPE and sufficient space or barriers to allow physical distancing.
When can I resume my community-based research?
In general, most community-engaged research protocols will fall into phase 4 research resumption. For guidance, see the Human Subject Research Phases Table. Much of community-engaged research involves face-to-face interaction or group activities; research teams that interact with individuals in community settings potentially pose a public health risk to both data collectors and to the communities in which they work.
In-person community-engaged research protocols should not resume unless standard safety recommendations can be implemented (social distancing, hand washing, mask wearing, and overall risk mitigation to the extent possible) and contact tracing is in place. For guidance, see the FAQ: What precautions should I take if I am conducting in-person research? Once research meets the phased criteria to resume, travel to and from community sites may be permitted, if compliant with university limitations/restrictions, interaction with members of the public is minimized, and the above safety recommendations are maintained. Even in phase 4, in-person research in which transmission risks cannot be fully controlled by the research team (e.g., research in public settings, participants’ homes, churches, etc.), will require pre-approval by the Office of the Vice President for Research.
What are some issues I should be aware of when resuming community-based research?
Community-engaged research protocols offer special challenges to the health and safety of research personnel and participants. While investigators must follow the same research resumption protocols as their non-community-engaged researcher colleagues (Department Chair/Center Director permission), guidance below is meant to assist generally to promote safety and well-being. For protocol-specific inquiries, contact ORI with the applicable protocol number and request an IRB consult to discuss resumption of research activities.
Investigators must determine appropriate local and community restrictions, requirements, and limitations pertinent to the conduct of the community-engaged research protocol. In an effort to prevent exposure of potential participants, research teams, and the general public to the COVID-19 virus, please follow any restrictions or requirements issued for/by the locations/facilities in which the human subjects research takes place.
If the research involves special circumstances or populations (e.g., minors in a school or other settings such as a community center), it is important to abide by the rules of the location but also make sure that parents and others who give consent are fully informed about risks and any measures taken to minimize those risks.
Unlike on-site research interactions, community-based research interactions may reach extensive networks of people, increasing the potential for rapid and mass exposure. Such exposure may make contact tracing with suspected or confirmed cases more challenging.
Research staff and participants may not know they are COVID-19 positive and could be carriers of the virus. For guidance on pre-screening potential research participants prior to in-person interaction, see the FAQ What precautions should I take if I am conducting in-person research?
When resuming community-engaged research activities, investigators and their teams are encouraged to provide educational resources to their participants. See the UK Community Resources for COVID-19 website.
What precautions should I take if I am conducting in-person research?
As in-person research interactions are permitted under the phases, researchers must consider how to modify in-person participant interactions to reduce the risk of COVID-19 transmission for both participants and the research team.
Researchers must:
- Develop robust testing protocols for staff testing, contact tracing, and self-isolation should a study team member test positive (along with reporting to the Kentucky Department of Public Health).
- Before coming to work, all faculty and staff are required to self-screen each day for signs of COVID-19 symptoms.
- Research staff who work at any UK Healthcare facility complete the UK Healthcare Staff Screening - https://covid-19.ukhc.org/employee-screening-for-covid-19/.
- Employees scheduled to work on-site but who are not feeling well and/or are experiencing any symptoms of illness must stay at home, and immediately contact their supervisor.
- Temperature checks may be performed prior to entering select research spaces or at discretion of investigator.
- Avoid all participants with confirmed or presumptive positive COVID-19 infection or who require potentially aerosolizing procedures, unless this is the focus of the study.
- Require the use of and supply Personal Protective Equipment (PPE). Address training status for nonclinical research personnel who need to be within 6 feet of participants, if appropriate for the study. Address sources of PPE for research use for staff and participants. Address methods to reduce PPE usage by all personnel, including combining research with clinical blood draws.
- Decrease potential exposure for participants and study staff at increased risk of severe illness from COVID-19. People at higher risk include people who:
- are over 60 years of age,
- have underlying health conditions including heart disease, lung disease, or diabetes,
- have weakened immune systems,
- are pregnant,
- are caregivers of children with underlying health conditions or adults who meet any of the criteria listed above.
Inform participants who may be vulnerable to contracting COVID-19 to review the
CDC guidelines for Groups at Higher Risk for Severe Illness. Enable participants to make an informed decision about personal risks related to COVID-19 and respect their wishes.
- Reduce face-to-face interactions with research participants and minimize the amount of time (e.g., < 15 minutes) of any required contact. Strategies may include physical barriers between research personnel and participants, or using technology (e.g., telephone, FaceTime, email, intercoms) to conduct interviews and obtain data.
- Continue remote telework for researchers or research staff whose responsibilities do not require on-site activities (e.g., regulatory specialist, administrative staff, survey researchers). Limit in-person research team meetings to those which allow personnel to maintain physical distancing procedures.
- Develop contingency plans in the event of staff illness or exposure.
- Close access to common areas where possible (e.g., waiting areas, staff break areas, etc.).
- Have a protocol for noncontact screening of participants and study staff for active symptoms of acute respiratory infection possibly related to COVID-19 or high risk of infection. This may be accomplished by communication with participants, care providers, chart review, screening surveys, or other options prior to approaching potential research participants. Individuals with active symptoms or in social isolation (e.g., due to travel history or exposure to someone with COVID-19 symptoms) should be avoided.
- Comply with UK and CDC guidance regarding sanitation of facilities and workspaces.
- Maintain targeted occupancy limits that permit physical distancing. This may require both physical separation of study personnel and staggered schedules and may require flexibility in work scheduling.
- Stagger participant visits so participants do not congregate at the research site and maintain at least six feet of separation between any participants who are present.
Researchers should also:
- Follow any guidelines or instructions from the specific facility (e.g., college, hospital, clinic) where the participant interaction would occur (e.g., screening).
- Consult with the study sponsor, when appropriate.
- Encourage remote monitoring by industry monitors or representatives. In-person sponsor visits may not occur until restrictions are lifted by local or institutional authorities.
Should I implement any special procedures in my research space to protect study personnel and research participants from exposure?
Please refer to the VPR Resumption of Research Phased Plan on personnel safety.
Identify the specific sources of COVID-19-related risks. Specific issues to consider include:
- Minimizing the number and duration (<15 minutes) of close interactions or actual physical contact between participants and your study team and/or other participants (e.g., a blood draw or physical exam) as much as possible.
- The feasibility of physical distancing and maintaining target occupancy limits.
- Use of physical barriers to reduce transmission risks.
- The availability of appropriate personal protective equipment (PPE).
- Screening before participant interactions.
- The necessary equipment or other items that will be used with multiple participants (e.g., a virtual reality headset, a computer-administered questionnaire, or a sensing device).
- What surfaces and objects will be touched by participants, including items at the research location (e.g., chairs, desktops, research equipment and devices, doorknobs).
- The availability and frequency of disinfection and cleaning procedures for frequently touched surfaces.
- Provision of adequate hand washing and sanitizing stations, etc.
- If the research procedures include testing participants for SARS-CoV-2 or serological antibodies, identify any restrictions (e.g., CLIA lab analysis) or requirements that apply (e.g., care referral and reporting for positive cases).
- All labs receiving COVID-19 patient specimens must have approval from the Institutional Biosafety Committee (IBC). Labs with current approvals for human source material will need to submit an amendment to their protocol. Registration information is available at https://ehs.uky.edu/Biosafety/ and protocols are submitted via https://topaz.uky.edu/. Email biosafety@uky.edu with questions regarding new protocol submissions or amendment creation.
Do I have to call to pre-screen all participants before they come in for a scheduled research visit?
Continued pre-screening is recommended consistent with your approved plan. For instance, rigorous pre-screening may be indicated for approval during Phase 4 to conduct research in the participant’s home or external facilities.
COVID-19 pre-screening calls do not require submission of a Modification Request as long as done for safety purposes and not research data collection. More details about screening are provided below.
What screening protocol should I use when interacting with research participants?
All research teams are advised to review and comply with facility, location, and/or department procedures for screening all participants.
Research teams are encouraged to initiate these screening procedures in advance of scheduled research visits and to cancel or reschedule visits for persons with current symptoms of a lower respiratory infection. Participants must “pass” screening to be seen in an academic or medical space.
In general, the following screening questions should be asked in advance of the scheduled visit:
- Has the participant had close contact with a confirmed COVID-19 positive individual for at least 15 minutes within the past 14 days? Close contact is defined by the CDC as:
- being within approximately 6 feet of a COVID-19 case for a prolonged period. Close contact can occur while caring for, living with, visiting, or sharing a health care waiting area or room with a COVID-19 case, or
- having direct contact with infectious secretions of a COVID-19 case (e.g., being coughed or sneezed on).
- Has the participant traveled to any locations confirmed by the Centers for Disease Control and Prevention to have widespread community transmission of COVID-19 in the past 30 days? See https://www.cdc.gov/coronavirus/2019-ncov/travelers/index.html for an updated list of areas with widespread community transmission.
- Does the participant have symptoms of a lower respiratory tract infection (e.g., cough, fever, sore throat, or shortness of breath, or loss of smell/taste) or gastrointestinal symptoms (e.g., diarrhea, nausea)?
If the answer to any of the questions above is “Yes,” terminate the visit and refer the participant to a medical facility for screening and care.
If interaction is taking place in a medical facility, follow medical facility directions on management of COVID-19 positive participants.
Screening outcomes:
- Document participant responses. If all is well, proceed with scheduling the visit and inform participant and/or caregiver regarding applicable visit procedures for on-site screening (e.g., registration screening; temperature checks), requirement to wear masks, and prohibition on bringing guests to the visit.
- If at least two weeks have passed since leaving an area of widespread transmission or exposure to a known COVID-19 case, and they have no symptoms of a lower respiratory tract infection, then the study visit may occur.
- If it is less than two weeks since their potential COVID-19 exposure and the participant has no symptoms, the study investigator should assess the potential risks and benefits of continuing the research visit versus rescheduling the visit until after the two-week period, if feasible.
- If the participant has symptoms of a lower respiratory tract infection, the investigator should consult the appropriate hospital resources to determine the next steps for evaluation and management.
Once participant is on-site, conduct screening procedures consistent with facility requirements, unless screening performed by facility staff upon entry or registration staff.
Are positive COVID-19 test results reportable to the state of Kentucky?
Yes, positive or presumptive positive SARS-CoV-2 results must be reported to public health authorities in the state of Kentucky. They may not accept results that come from a lab which is not certified by the federal CLIA program or state equivalent. If you obtain a positive result, report it to the state of Kentucky and, if required, UK HealthCare. If the test is not a recognized/verified test, you should tell the participant about the result and the limits of that test, then encourage them to get a valid test from a healthcare facility. Find instructions under the “Healthcare Providers” section under “Resources” on the Kentucky Public Health COVID-19 website: https://govstatus.egov.com/kycovid19.
Guidance from the Office for Human Research Protections (OHRP) Guidance on COVID-19 states: “Legally Required Reporting: When required by law to provide information related to an individual's COVID-19 test results to a public health authority, including individually identifiable information about individuals who are research participants, the HHS protection of human subjects regulations do not prevent investigators or institutions from fulfilling this requirement (even if doing so would be inconsistent with statements made in the study's consent form). The existence of a Certificate of Confidentiality[3] does not alter an investigator's ability to disclose a research participant's COVID-19 test results when required by federal, state, or local laws. For example, if a research participant tests positive for COVID-19, an investigator may provide this test result to a public health authority if required to do so under applicable state or federal law. In such circumstances, investigators should inform the participant of the required reporting of results.” [https://www.hhs.gov/ohrp/regulations-and-policy/guidance/ohrp-guidance-on-covid-19/index.html]
For Questions About Screening, Testing or Tracing? Contact Our Health Corps.
Call 859-218-SAFE(7233)
Email healthcorps@uky.edu
Hours of Operations:
Monday-Friday 8 a.m. - 8 p.m.
Saturday-Sunday 10 a.m. - 5 p.m.
After research reopens, if a subject or research team member is discovered to have tested COVID positive or presumptive positive with 2 weeks of an in-person research visit, would we promptly report to the IRB as an Unanticipated Problem involving Risks to Subjects or Others (UPIRSO)?
An Unanticipated problem involving risks to subjects or others (UPIRSO) - includes any incident, experience, or outcome that meets all of the following criteria:
- Unexpected (in terms of nature, severity, or frequency) given (a) the research procedures that are described in the protocol-related documents, such as the IRB-approved research protocol and informed consent document; and (b) the characteristics of the subject population being studied;
Consider whether unexpected in frequency given the presumed measures that in place to mitigate risks. Might be expected for COVID-19 research. - Related or possibly related to participation in the research; and
Could it have occurred because of an in-person research interaction/intervention? Likely if interaction is for research and not concurrent with other interaction. - Suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized.
Answer: Likely so since a positive result (including presumptive positive) suggests others are at greater risk of virus transmission than was previously known or recognized.
What do I need to know to submit/conduct COVID-19 research?
The document below includes information on submitting new or updating existing research which focuses on COVID-19.
Conduct and Submission of COVID-19 Research in Human Subjects [PDF]
What is the PREP Act and what should subjects in COVID-19 interventional trials be told in the informed consent about liability limits?
On March 10, 2020, the HHS Secretary issued a Public Readiness and Emergency Preparedness (PREP) Act Declaration which was amended effective March 27, 2020 that provides liability immunity (except for willful misconduct) for claims of loss caused, arising out of, relating to, or resulting from manufacture, distribution, administration or use of countermeasures to diseases, threats, and conditions determined by the Secretary to constitute a public health emergency to entities and individuals involved in the development, manufacture, testing, distribution, administration, and use of such countermeasures.
PREP limits legal rights to sue covered persons engaged in select COVID-19 countermeasures (including select treatment research) and provides compensation for eligible individuals who suffer injuries from use of covered products.
Covered Countermeasures may include vaccines, drugs, or medical devices to be used to treat, diagnose, cure, prevent, or mitigate COVID-19.
Products must be:
- a qualified pandemic or epidemic product;
- a security countermeasure;
- a respiratory protective device approved by NIOSH;
- approved, licensed, or cleared by FDA;
- authorized under an Emergency Use Authorization (EUA) issued by FDA;
- described in an Emergency Use Instructions (EUI) issued by the CDC; or
- used under either an Investigational New Drug (IND) application or an Investigational Device Exemption (IDE).
In addition to meeting the scope of the act, reasonable precautions must be taken to facilitate the safe use of covered countermeasures. See the PREP Act Q&As and PREP Act Glossary for detailed information.
CONSENT LANGUAGE:
The following statement should be added to the informed consent for applicable COVID-19 studies, in order to inform participants regarding their legal rights.
Due to the coronavirus public health emergency, the federal government has issued an order that may limit your right to sue if you are injured or harmed while participating in this COVID-19 study. If the order applies, it limits your right to sue and recover losses from the researchers, healthcare providers, any study sponsor, distributor or manufacturer involved with the study. However, the federal government has a program that may provide compensation to you or your family if you experience serious physical injuries or death. To find out more about this “Countermeasures Injury Compensation Program” please go to https://www.hrsa.gov/cicp/about/index.html or call 1-855-266-2427.
If participants have questions about the Countermeasures Injury Compensation program (CICP) direct them to the CICP website which provides Requester FAQs, Fact Sheet and contact information.
Are there other resources I can view for further guidance?
- UK Vice President for Research (VPR) COVID-19 Guidance for Researchers https://www.research.uky.edu/resources/covid-19-guidance-researchers
- UK Institutional Biosafety COVID-19 Guidance - https://ehs.uky.edu/biosafety/ (Scroll down to COVID-19)
- UK Healthcare COVID-19 Website - https://covid-19.ukhc.org/ includes guidelines, TeleCare Toolkit, FAQs, Infection Prevention and Control, UKHC Employee Screening and Resources.
- UK Healthcare Staff Screening - https://covid-19.ukhc.org/employee-screening-for-covid-19/
- UK COVID-19 FAQs - https://www.uky.edu/coronavirus/faqs
- Employee and Human Resources Information - https://www.uky.edu/coronavirus/faqs#hr
- Campus Restart - https://www.uky.edu/coronavirus/faqs#restart
- Kentucky Public Health - https://govstatus.egov.com/kycovid19 (Scroll to RESOURCES for Guidance by Topic and Healthcare Provider Information on Case Reporting)
- Kentucky COVID-19 hotline provides guidance for individuals in the state of Kentucky with symptoms 800-722-5725
- Centers for Disease Control and Prevention Coronavirus website https://www.cdc.gov/coronavirus/2019-ncov/index.html
- Council on Governmental Relations (COGR) Research Ethics & Compliance Committee - Human Subjects FAQs May 4, 2020 (Version 1.0)
https://www.cogr.edu/sites/default/files/FAQS%20ON%20HUMAN%20SUBJECTS%20for%20REC%20final%20may%204%202020.pdf - Food and Drug Administration (FDA) "Assessing COVID-19-Related Symptoms in Outpatient Adult and Adolescent Subjects in Clinical Trials of Drugs and Biological Products for COVID-19 Prevention or Treatment" https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-covid-19-related-symptoms-outpatient-adult-and-adolescent-subjects-clinical-trials-drugs
- "FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency" (Updated September 21, 2020)https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-guidance-conduct-clinical-trials-medical-products-during-covid-19-public-health-emergency
- Food and Drug Administration (FDA) "FDA Digital Health Policies and Public Health Solutions for COVID-19"
https://www.fda.gov/medical-devices/digital-health/digital-health-policies-and-public-health-solutions-covid-19 - National Cancer Institute Central IRB (NCI-CIRB) "Memorandum On Interim Guidance For Patients On Clinical Trials Supported By The NCI Cancer Therapy Evaluation Program And The NCI Community Oncology Research Program (NCORP)"
https://www.ncicirb.org/announcements/memorandum-interim-guidance-patients-clinical-trials-supported-nci-cancer-therapy - National Cancer Institute Central IRB (NCI-CIRB) "Interim Guidance for Patients on Clinical Trials Supported by the NCI Cancer Therapy Evaluation Program and the NCI Community Oncology Research Program (NCORP)" (March 13, 2020)
https://www.ncicirb.org/announcements/memorandum-interim-guidance-patients-clinical-trials-supported-nci-cancer-therapy - National Cancer Institute Central IRB (NCI-CIRB) "Additional Guidance Regarding Alternative Procedures for Clinical Trials Supported by the NCI Cancer Therapy Evaluation Program(CTEP) and NCI Community Oncology Research Program (NCORP) Affected by the Spread of the Novel Coronavirus" (March 23, 2020)
https://ncicirb.org/sites/ncicirb/files/Memo_Additional_Guidance_Clinical_Trial_Activities_Affected_Coronavirus.pdf - National Institutes of Health (NIH) has issued notices on the NIH Extramural Response to Natural Disasters and Other Emergencies website
https://grants.nih.gov/grants/natural_disasters.htm - Office for Civil Rights (OCR) "Notification of Enforcement Discretion for Telehealth Remote Communications During the COVID-19 Nationwide Public Health Emergency"
https://www.hhs.gov/hipaa/for-professionals/special-topics/emergency-preparedness/notification-enforcement-discretion-telehealth/index.html - Office for Human Research Protections (OHRP) Guidance on COVID-19 https://www.hhs.gov/ohrp/regulations-and-policy/guidance/ohrp-guidance-on-covid-19/index.html
What if my research involves a public health authority or public health surveillance?
If you think your work is public health surveillance in addition to (or instead of) research, please fill out a UK Not Human Subjects Research (NHR) form after obtaining documentation (e.g., email) that describes the specifics of any involvement of a public health authority. You should include the following in the summary of the NHR form:
- a brief description of the role of the public health authority (e.g., design, coordination, direction);
- clarification of whether the public health authority plans to use the data to make decisions; and
- if so, when and for what purpose the data will be used.
This information is important because it impacts the type of IRB review required (if any).
What public health surveillance activities are deemed not to be human subject research under the revised Common Rule?
- The activity must be a public health surveillance activity (45 CFR 46.102(l)(2));
- The activity must be conducted, supported, requested, ordered, required, or authorized by a public health authority (45 CFR 46.102(k) and 46.102(l)(2)); and
- The activity must be limited to that necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance (including trends, signals, risk factors, patterns in diseases, or increases in injuries from using consumer products) (45 CFR 46.102(l)(2)).
However, FDA regulations may apply if this involves use of an investigational in-vitro diagnostic device.
Detailed guidance is available from the Office for Human Research Protections (OHRP).
The following provides guidance for human research regarding studies:
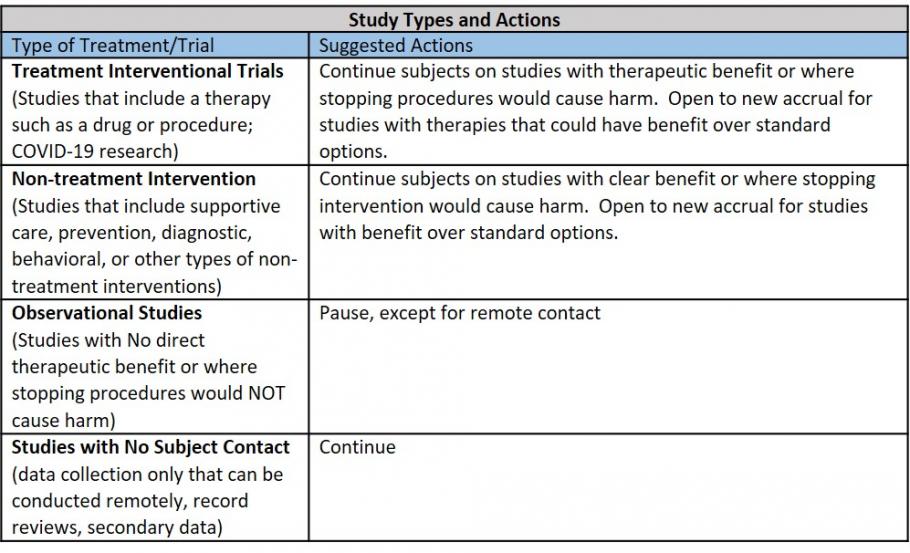
Which human subject research studies should be paused during the COVID-19 pandemic?
Studies that involve contact with study subjects but have no direct benefit to the subject should be paused until further notice.
Examples of paused studies include:
- Studies collecting human samples or imaging that require study subject contact (e.g., phlebotomy, surveillance biopsy, radiographic imaging, physical examination) without direct diagnostic or therapeutic benefit;
- Interview studies that can’t be conducted remotely;
- Observational studies involving group gatherings;
- Community studies.
Examples of studies not included in the pause:
- Clinical trials with treatment arms (e.g., oncology clinical trials, interventional studies for devices or therapies with significant benefit to subjects). For purposes of the pause, it is assumed that trials with investigational treatments, including drugs and devices, provide the potential for benefit and should continue. Investigators should weigh the benefits of the treatment arm against the risks of COVID-19 exposure for study staff or research subjects as they make a decision about continuing these projects;
- COVID-19 studies, observational or interventional;
- Data collection only that can be conducted remotely (e.g., EMR, previously collected) Studies that do not involve any subject contact (e.g., UK Healthcare Zoom, UK Telehealth, REDCap). Refer to UK IT Cybersecurity Best Practices When Working Remotely. (Updated 3/30/20)
Can I submit a new research project?
Yes. The IRB will continue to review new study submissions through E-IRB.
Should I indicate if my research project is related to COVID-19 in my project title?
***Effective April 16, 2020: If your research involves investigating any aspect of COVID-19, please enter “COVID-19” at the start of your Project Title in your IRB protocol application.***
Please add COVID-19 to “Title of Project” and “Short Title Description” of your current IRB protocol.
For protocols being reviewed by an external IRB, please enter “RELIANCE COVID-19” before the PROJECT and SHORT titles.
Is there a biobank for UK Researchers specifically for research on COVID-19?
Yes, an announcement from CCTS on April 23, 2020 states:
In response to the COVID-19 pandemic, the University of Kentucky Center for Clinical and Translational Science (CCTS) has created a new biospecimen bank to support research on the novel virus. Logistical support for the biobank is led by the UK Markey Cancer Center.
The COVID-19 Research Registry and Specimen Bank will collect specimens from volunteers who have tested positive for or are suspected of having the virus. The bank will work closely with the UK CURE (COVID-19 United Research Experts) Alliance, a new workgroup within UK’s College of Medicine that unites experts from across campus to focus on advising COVID-19 patient care and clinical trials.
Samples can be requested through the CCTS Service Request Form by selecting "Biospecimens" and then "COVID-19 Biobank."
Is an IRB Modification Request required to delay, pause, or temporarily modify research activities due to COVID-19?
Research protocols or any single component of a research protocol may be delayed/paused or temporarily modified for COVID-19 concerns without submitting a Modification Request to the IRB. Per federal regulations, an investigator can make changes to a protocol to eliminate apparent hazards to research participants without first obtaining IRB approval. The change may apply to a single participant or all participants enrolled in the research study.
However, if changes to research procedures are permanent or propose new COVID research and not currently approved by the IRB, a Modification Request should be submitted prior to implementing the changes.
For FDA studies, please see next question.
Does FDA require sites to track and report changes that were made without IRB approval to minimize hazards from COVID-19?
Yes. Changes to protect the life and well-being of research subjects (e.g., to limit exposure to COVID-19) may be made without IRB approval, but FDA-regulated studies require such changes to be reported afterwards. Investigators should submit an aggregate account of changes implemented without prior IRB approval on one protocol violation report.
FDA has advised sponsors to collect a list of contingency measures taken; list subjects affected by study number; and indicate the impact on safety or efficacy results, if applicable. Check with the study sponsor on reporting requirements.
Clinical trials that have paused recruitment due to COVID-19, should update their study status on ClinicalTrials.gov record to “active, not recruiting” within 30 days. For questions regarding ClinicalTrials.gov, please contact Emily Bradford at emily.bradford@uky.edu.
Do I need to notify the IRB to pause recruitment?
No. This is not a change to the protocol, but a temporary pause in enrolling additional subjects, so notification is not required. You do need to notify the study sponsor. You should document the pause in your study files.
- For UK IRB approved research: If study visits can be conducted virtually (e.g., UK Zoom, or telephone), the study teams should document any missed assessments or items that could not be performed because it was a remote visit.
They should not submit modifications to add this possibility of remote research visits, as we expect this to be a temporary situation. We will provide further guidance about how and when to submit this documentation. - For research approved by another IRB: Please contact that IRB for their instruction.
How should I manage remote communications or data collection with subjects?
Privacy and confidentiality provisions remain critically important at all times, even when working remotely. Please note, collection, transmission, or access to private identifiable data or protected health information, must comply with university and other policies for security of research data.
Remote interactions that will collect or transfer private identifiable the information or protected health information should use technology that is IT secure or HIPAA compliant (e.g., UK Healthcare Zoom, UK Telehealth, REDCap). UK Healthcare has a HIPAA compliant contract with Zoom for secure videoconferencing. Link to ukth.zoom.us to set up and account with your linkblue ID and password. Refer to UK IT Cybersecurity Best Practices When Working Remotely.
Do not store private identifiable information or protected health information on unsecure devices in order to work remotely. Use University-approved cloud services and VPN access while working at home instead of storing data directly on personal devices.
If making copies of physical research records or data (paper consent forms, case report forms, questionnaires/surveys, etc.), secure all paper locally following HIPAA principles and return to the IRB-approved location as soon as is practical.
See additional tips for remote security https://www.totalhipaa.com/hipaa-compliance-working-remotely/
Do I need to notify the IRB if all study activities are paused?
Not at this time. You must notify the study sponsor (see below). Please track when you pause all study activities and document all missed visits, procedures, appointments, and other relevant study activities.
Do I need to re-consent subjects on changes to protect from COVID-19?
In most cases, new information regarding changes to protect from COVID-19 may be provided via a letter or other form of communication. Subjects must be provided with significant new findings that develop during the research which may relate to their willingness to continue participation. However, re-consent is not necessary unless the changes to the research are such that the original consent is no longer valid. See the ORI New Information Guidance for help determining whether notification is adequate for providing new information or re-consent is warranted.
What guidance is available for e-consent or remote consent?
Remote informed consent may be proposed for new COVID-19 research or implemented for an existing protocol in response to the pandemic. Electronic consent may be proposed for in-person or remote consent. The ORI Outline of FDA Guidance for Industry: Use of Electronic Informed Consent in Clinical Investigations summarizes federal guidance and consideration of system security, identity verification, legally valid e-signatures, and the provision of subject copies.
For options conducting remote consent for subjects who cannot travel or patients in isolation, see the UK ORI Remote Informed Consent Guidance or questions 10 & 11 in the FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency.
The University of Kentucky Center for Clinical and Translational Science (CCTS) has presented a REDCap mobile app and consent feature webinar. In addition, they have shared a video demo of their process for obtaining remote consent of UK COVID-19 Biobank participants using REDCap.
What if I have additional questions about my study?
For Clinical Research questions: You may contact the Center for Clinical and Translational Sciences by email at CCTS@uky.edu or phone at (859) 323-3775 and your question will be forwarded to a content expert who will try to answer your question.
For General IRB or Human Research questions: You may contact the Office of Research Integrity by email at IRBSubmission@uky.edu or phone at (859) 257-9428 and your question will be forwarded to the appropriate staff member. If your question is about a particular study, please include the IRB protocol number and PI name.
What e-consent options are FDA Part 11 compliant for FDA-regulated clinical trials?
Software may not be compliant with FDA Part 11 out of the box. It may need to be configured and processes should be implemented to ensure use is in a manner that meets Part 11 requirements, including identity verification, signature certification, and audit trails. Below is information and resources on potential options. Note: this does not constitute approval or endorsement of the IRB. Also, the customer retains responsibilities for implementation of features, functions, and training in order to meet regulatory compliance, implemented features and processes.
DocuSign
Docusign’s Part 11 module website provides an overview and operational information on administration, settings, and features. DocuSign's Part 11 module contains industry-designed capabilities that include:
- Pre-packaged account configuration
- Signature-level credentialing
- Signature-level Signing Reason
- Signature manifestation (Printed Name, Time Stamp, and Signature Reason)
- Detailed audit trail
- Tamper-evident digital seal
Adobe Sign
Adobe Sign is a cloud-based e-signature service which supports electronic and digital signatures. Consent documents may be uploaded into the Adobe Sign portal and sent by email to prospective participants. For each feature, the Adobe Sign and 21 CFR Part 11 guide, prepared by Montrium Consulting, describes how it complies and the associated shared customer responsibilities.
FDA COVID MyStudies Application (App)
FDA has made previously developed platform to obtain informed consent securely when face-to-face contact is not possible or practical due to COVID-19 control measures. Investigators download COVID MyStudies in the Apple App and Google Play. The app may be used to send informed consent documents to individuals or LARs. Investigators and participant receive copies of completed digital informed consent documents.
Does UK provide general guidance for study subjects, in addition to that provided by individual study staff?
Yes. The UK Center for Clinical and Translational Science (CCTS) provides general Guidance for Research Participants during COVID-19.
What guidance does FDA provide for clinical trials during the COVID-19 Pandemic?
The FDA outlines considerations to assist sponsors in assuring the safety of trial participants, maintaining compliance with good clinical practice and minimizing risks to trial integrity. Considerations recommended include, among others, sponsors evaluating alternative methods for assessments, like phone contacts or virtual visits and offering additional safety monitoring for those trial participants who may no longer have access to investigational product or the investigational site.
For more information visit:
"FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency" (Updated September 21, 2020)
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-guidance-conduct-clinical-trials-medical-products-during-covid-19-public-health-emergency
Other FDA Guidance:
"Assessing COVID-19-Related Symptoms in Outpatient Adult and Adolescent Subjects in Clinical Trials of Drugs and Biological Products for COVID-19 Prevention or Treatment"
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-covid-19-related-symptoms-outpatient-adult-and-adolescent-subjects-clinical-trials-drugs
"FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency"
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-guidance-conduct-clinical-trials-medical-products-during-covid-19-public-health-emergency
Has the NIH issued any formal guidance on clinical trials or other human subjects research activities?
Yes, although at this point in time the guidance is broad and geared predominately at administrative issues.
Guidance can be found on the NIH web page for Natural Disasters:
https://grants.nih.gov/grants/natural_disasters.htm.
What is the suggested COVID-19 screening protocol for study subject visits?
Call every subject the day before a site or home visit and ask the following travel screening questions:
- Have you or your family members traveled in the last 14 days?
If yes, ask for dates and locations of travel. If travel was outside of the local upstate region you may elect to postpone the visit by two weeks. - Do you or anyone in your home have any of the following symptoms: cough, fever, shortness of breath? Have you or anyone in your family had any contact with a confirmed COVID-19 patient?
If yes to either question, then postpone the in-person visit for at least two weeks and make sure they have spoken with a health care professional. - All subjects should be re-screened when they arrive for their visit.
If a subject presents with the symptoms listed above at their study visit, terminate the visit and have them contact their primary provider.
What should I do about missed visits for trials that remain open?
If a subject misses a scheduled visit during this period, it should be documented including the reason for the missed visit (ex. COVID-19). Contact the study sponsor for guidance on how they would like such notifications and documentation handled. It is likely, given that this will be common during this national emergency, that notification requirements will be given to all study sites.
What should I do to prepare for study subjects being removed from active protocols due to COVID-19 infection?
Review each clinical trial protocol for drug reductions or holds due to serious adverse events or for other reasons. Subject who are diagnosed with COVID-19 will likely be removed from study treatment by the sponsor or investigator. Treatment schedule modifications or removal from the study for subjects who are in quarantine should be discussed with the sponsor. Items should be reported consistent with the UK IRB reporting policies.
Should I modify my study protocol to reduce risk to my study subjects?
When possible, you should consider such modifications.
Eliminating immediate risks may include actions to reduce potential exposure to COVID-19 or to continue to provide medically necessary study care (including study drug) to subjects who have been placed in isolation or quarantine because of suspected or known exposures. We encourage investigators to take necessary steps to eliminate additional risks to subjects. Even in studies that offer benefit to the patient, investigators should balance the potential benefits with the risk of patient exposure to COVID-19 by further participation.
Depending on the protocol, exposure or a positive test for COVID-19 could be classified as an adverse event, and could also cause a protocol-required reduction or temporary or permanent discontinuation of investigational drug. Giving study drug to isolated or quarantined subjects will also be situational. For example, IV infusions could pose a serious problem if given outside the clinic. The protocols will often indicate under which conditions drugs should be discontinued and how. Death during a study is a serious adverse event.
We recommend that study teams communicate with sponsors/clinical research organizations about these scenarios and document the responses.
What if the sponsor issues new guidance or information about COVID-19 risks for my study?
You will need to submit a modification request to the IRB. Your study may be paused if the sponsor, Data Safety Monitoring Board (DSMB) or you identify any new increased risk to study subjects related to the protocol.
What should I do about study/sponsor monitor visits? (Updated 8/26/20)
To the extent possible, all monitoring visits, site initiation visits, site qualification visits, should occur remotely.
Otherwise, communicate facility safety requirements to the study sponsor and/or clinical research organization, incorporate barriers or social distancing when making site-visit arrangements, and perform safety screening of on-site monitors prior to visits.
Document the contact in the study documentation.
What facilities exist for virtual visits with study subjects or study monitors?
Depending on the recipients' resources, communications options include telephone conferencing, UK Zoom, UK Telehealth, REDCap, and other survey platforms.
The Center for Clinical and Translational Science (CCTS) provides a procedure for obtaining view-only access to the Electronic Medical Record (EMR) and offers options for monitoring documents outside of the EMR using existing platforms at UK (One Drive and Zoom). EMR access is enabled by UK HealthCare IT and is subject to review and/or audit by the Office of Corporate Compliance. Details and resources for remote monitoring are available at ccts.uky.edu/RemoteMonitoring.
Are ORI Quality Assurance Wellness Checks being conducted at this time?
The ORI Quality Improvement/Quality Assurance (QA/QI) Program is not scheduling or conducting any visits at this time due to COVID-19 concerns. We will resume our visits once the university returns to regular business function and the conduct of in-person events.
Please contact Pam Stafford pastaf3@uky.edu if you have questions or concerns.
Are IRB meetings being conducted remotely?
Until further notice, IRB meetings will continue to be conducted electronically through Zoom. Investigators will receive an invite containing a Zoom link prior to the meeting.
Investigators should inform ORI staff if they have other study personnel to whom they need a meeting invitation sent. Investigators should not forward the meeting link to their study personnel. It’s important for ORI staff to know everyone who will be attending so they are aware of who is and is not supposed to be admitted into the virtual meeting during each scheduled appointment time.
In addition, the number of attendees for each protocol needs to be limited in order to keep attendance tracking manageable for documentation purposes, so we ask that investigators have no more than two (2) individuals from the list of study personnel join them for the meeting.
Announcements will be made in advance of any further changes to in-person or hybrid meetings.
What if my protocol was reviewed by an external IRB?
Check with reviewing IRB on reporting requirements as they may differ with local IRB
Phase 3:
The following FAQs provide Phase 3 specific information that is supplemental to the Human Subjects Research Phases Table.
Please read these FAQs before submitting an Individual PI Plan to resume or initiate Phase 3 research.
What general considerations apply for human subjects research which may qualify to resume during Phase 3?
The same guiding principles of social distance/physical barriers, appropriate personal protective equipment (PPE), and performance of remote human subjects research activities, whenever possible, remain as first-choice options. In addition, the VPR Phased Resumption Plan includes Key Points for Minimizing COVID-19 exposure and transmission at all phases. PIs should consider revising their protocols to minimize the number of research personnel, limit the time of exposure, revise use of PPE, and any other measures that reduce the possibility of infectivity.
Community, societal, and scientific benefit must be justified and be measured against risk of research participant exposure to COVID-19. Each potential point of transmission risk should be carefully considered and detailed in the investigator’s plan, including clear descriptions of the ways that risk will be mitigated/controlled. Studies that cannot adequately mitigate risk of transmission cannot be conducted until Phase 4.
What specific restrictions or requirements are there for Phase 3 research?
Since Phase 3 research does not offer direct benefit to the individual human subject, the following restrictions or requirements should be employed:
- Research with individuals who are “at risk” (e.g., elderly, pre-existing conditions) in which participation does not offer direct benefit to the individual should not take place until Phase 4.
- In-person interaction involving physical contact, should be limited to a brief period (e.g., 15 min as recommended by the CDC) with a single participant, minimal staff present and employing the highest level of safety precautions (e.g., N95 masks, gowns, gloves, eye protection or face shields or barriers). Brief interactions are less likely to result in transmission of virus. Thus, decisions should be made consistent with current CDC recommendations (https://www.cdc.gov/coronavirus/2019-ncov/php/public-health-recommendations.html), as there is growing evidence of transmission risk before onset of symptoms.
- Generally, research conducted inside the home of an individual human participant is not advisable as mitigation of risks cannot be controlled.
- Research with in-person group meetings of 10 or more should be conducted outdoors or in facilities allowing a minimum 6-foot social distancing or physical barriers and adequate air exchange.
- Regarding procedures involving aerosolized saliva, the American Thoracic Society recommends that pulmonary function testing be limited only to cases that are essential for immediate treatment decisions, and that measures to protect both the staff and individuals being tested should be put in place. Research procedures that result in aerosolizing saliva include, for example, sputum induction, pulmonary function tests in nonclinical space, sputum culture, pulmonary exercise tests, vigorous exercise resulting in increased and/or forceful respiration, and singing. Such research should not take place until Phase 4, unless the following protective measures are implemented: personal protective equipment (PPE) that limit aerosolized droplet acquisition for staff (mask, eye protection, shield, gown, gloves), and enhanced sanitizing of the testing space. Other in-door research procedures or activities that result in aerosolizing saliva (e.g., vigorous exercise, singing) will need to implement similar protective measures or the research should be delayed until Phase 4.
In transitioning to Phase 3, are there specific human participants research populations that warrant additional safeguards or support?
During human interaction, researchers should apply universal precautions as anyone is a potential carrier of COVID-19. However, the CDC has defined populations that may need extra support, access, or precautions (e.g., homeless people, people with disabilities, refugees, etc.). Population specific guidance is available at https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/index.html.
Can undergraduates serve as research personnel in human participant research studies that take place during Phase 3?
Consistent with the VPR Plan (Section D), high school students are not allowed to serve as study personnel on research protocols including human participants during Phase 3. Generally undergraduates serving as study personnel on research protocols with human participants should only be permitted in cases with minimal incidental risk. Undergraduates are prohibited from participating in COVID-related research, but exceptions will be considered if risk is adequately mitigated. In order to maximize health and safety during Phase 3, both justification and a description of risk mitigation is required in the PI plan for specific undergraduates to serve as research personnel in research protocols with human participants. Please include the name and contact information for the undergraduates in the PI plan. Prioritization should be given to undergraduates taking research course credits for degree completion and to undergraduates in paid research positions.
Please note: you must meet the requirements of the VPR Resumption of Research Phased Plan (training & individualized plan submission for Phases 2-4) before conducting any human subjects research. Both the researcher training completion and the individualized plan for research work space/personnel are automatically routed by the VPR’s office to the respective department chair for evaluation. Unless otherwise indicated, protocol-specific IRB approval to resume research that has been paused due to COVID-19 concerns is not required.
Phases for Resumption of Human Subjects Research
-
When and how will human subject research be permitted to resume?
-
What is the current status of human subjects research at UK? (Updated 8/26/20)
-
How do I determine if I can conduct in-person research? (Updated 6/10/20)
-
Human Subjects Research Phases Table
-
Once I am permitted to resume the paused research, will there be any restrictions on locations?
-
To whom do I submit a request to resume the paused research?
-
What if my research does not involve or require in-person contact?
-
What if a study without direct participant benefit permits some, but not all, interactions to coincide with existing events where the participant is already at the facility? (Updated 6/2/20)
-
What if I’m conducting a comparative, survey, or observational study that does not have direct participant benefit, but the in-person interactions align with existing visits or in-patient admissions?
-
What if my research study is included in the “allowed” category, but I am not able to comply with precautions and safety guidelines related PPE, physical distancing, and maximum occupancy in a facility?
-
When can I resume my community-based research?
-
What are some issues I should be aware of when resuming community-based research?
-
What precautions should I take if I am conducting in-person research?
-
Should I implement any special procedures in my research space to protect study personnel and research participants from exposure?
-
Do I have to call to pre-screen all participants before they come in for a scheduled research visit?
-
What screening protocol should I use when interacting with research participants?
-
Are positive COVID-19 test results reportable to the state of Kentucky?
-
After research reopens if a subject or research team member is discovered to have tested COVID positive or presumptive positive with 2 weeks of an in-person research visit, would we promptly report to the IRB as an Unanticipated Problem involving Risks to Subjects or Others (UPIRSO)?
-
What do I need to know to submit/conduct COVID-19 research? (Added 6/1/20)
-
What is the PREP Act and what should subjects in COVID-19 interventional trials be told in the informed consent about liability limits? (Added 6/1/20)
-
Are there other resources I can view for further guidance? (Updated 9/22/20)
-
What if my research involves a public health authority or public health surveillance?
-
What public health surveillance activities are deemed not to be human subject research under the revised Common Rule?
When and how will human subject research be permitted to resume?
Paused human subjects research will resume in phases along with the Vice President for Research (VPR) Resumption of Research Phased Plan and the Governor’s Healthy at Work Reopening Plan. These phases are based on environmental factors beyond the control of UK/UK Healthcare and, therefore, dates (even tentative) cannot be provided.
In light of the UK phases for resuming research, considerations when resuming human subjects research may be viewed as conservative. However:
- Research participants are volunteers.
- Many studies do not provide direct benefit to individual participants.
- It is difficult to ascertain risks due to COVID-19 at a location because there are so many variables.
- It is essential to monitor and control the number of studies being conducted as we resume research to ensure appropriate occupancy and safe management of resources (e.g., research space, staffing densities, and the availability and use of PPE).
What is the current status of human subjects research at UK? (Updated 8/26/20)
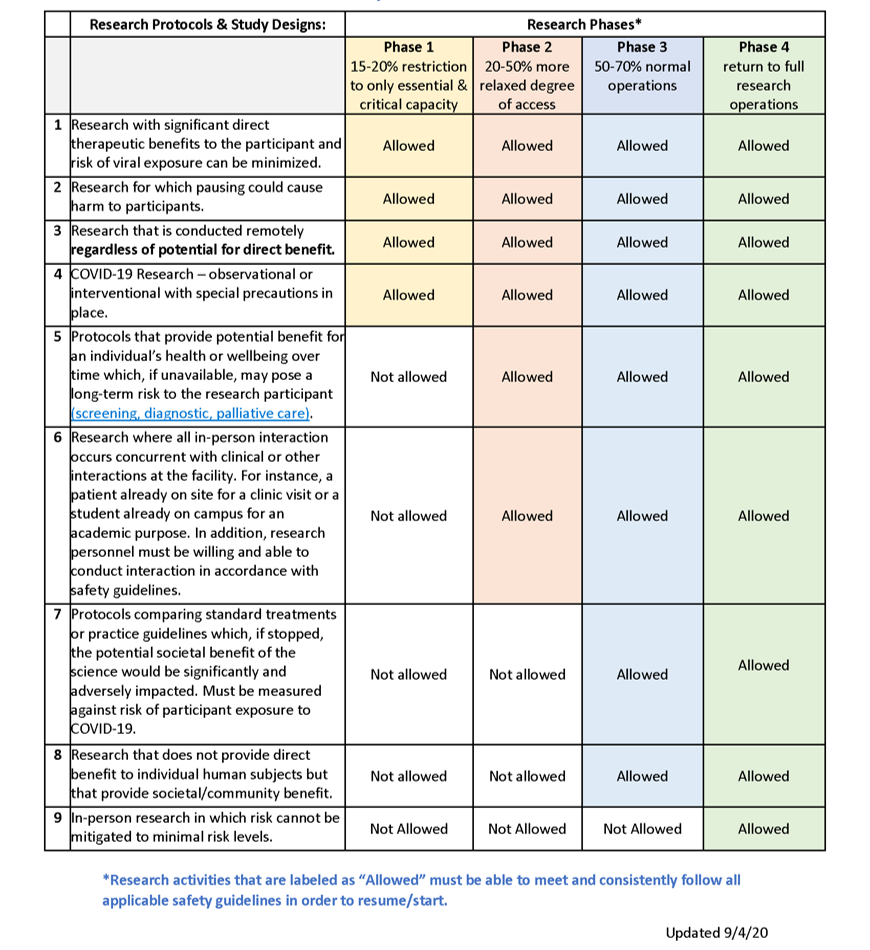
Please refer to the VPR’s University of Kentucky Resumption of Research Phased Plan for guidance on the current phase and the ORI Human Subjects Research Phases Table to determine if your human research is allowed to resume in the current phase after meeting all requirements outlined below.
How do I determine if I can conduct in-person research? (Updated 6/10/20)
This determination is a combination of:
- Meeting the requirements outlined in the VPR Resumption of Research Phased Plan (i.e., waiver for Phase 1, training & individualized plan submission for Phases 2-4);
- Compliance with the Human Subjects Research Phases Table; and
- Permission from your Department Chair or Center Director.
The phased approach and these guidelines provide a baseline framework from which additional variables must be considered.
- Continuing or resuming human research is contingent on active COVID-19 cases, available testing and contact tracing, and compliance with health authority guidance from national, state, local, and University authorities.
- Polices and guidance will be dynamic and may accelerate or decelerate based on shifting information and incidence data. Researchers must be prepared to halt activities on short notice if this becomes necessary based on new information.
- Resuming human research operations must be parallel to and consistent with the institutional and VPR’s plans, as well as specific facility, college, and/or unit mandates.
Investigators may choose to be more stringent or cautious than phases allow.
It is critical to keep research participants informed of study status and changes that could impact them. Respect participant wishes regarding resuming in-person interaction.
Human Subjects Research Phases Table
Once I am permitted to resume the paused research, will there be any restrictions on locations?
Please refer to the VPR Resumption of Research Phased Plan for UK locations. For other research locations/facilities, determine whether those locations/facilities have any restrictions or requirements for human subjects research.
To whom do I submit a request to resume existing human subject research?
Submit your plan to resume human subjects research, using the VPR template, for review by your Department Chair or Center Director.
What if my research does not involve or require in-person contact?
Research that does not involve in-person contact (e.g. record review, secondary data analysis) or studies modified to fully remote interactions with participants or staff were not paused and may continue.
What if a study without direct benefit permits some, but not all, interactions to coincide with existing events where the participant is already at the facility? (Updated 6/2/20)
Unless a study provides potential direct benefit or long-term risk if not resumed, all research-only in-person interactions must be changed to either remote visits or be rescheduled to coincide with existing events/encounters where the participant will already be on site. Research-only in-person interactions for studies which do not offer potential individual benefit will be allowed to resume at stage 3 or 4 respectively.
What if I’m conducting a comparative, survey, or observational study that does not have direct participant benefit, but the in-person interactions align with existing visits or in-patient admissions?
In general, these studies can only resume at Stage 3. The intent is to limit and control the number of studies that are being conducted at one time in order to maintain safe space densities and PPE inventories.
What if my research study is included in the “allowed” category, but I am not able to comply with precautions and safety guidelines related PPE, physical distancing, and maximum occupancy in a facility?
Research must be included in the allowable category AND meet safety guidelines to resume operations. This includes PPE and sufficient space or barriers to allow physical distancing.
When can I resume my community-based research?
In general, most community-engaged research protocols will fall into phase 4 research resumption. For guidance, see the Human Subject Research Phases Table. Much of community-engaged research involves face-to-face interaction or group activities; research teams that interact with individuals in community settings potentially pose a serious public health risk to both data collectors and to the communities in which they work. Thus, while ramping up research activities (for example, scheduling future events or assembling research materials on-site) may begin to take place in phase 3, resumption of such interaction necessitates waiting until phase 4. For guidance, see the FAQ: How do I determine if I can conduct in-person research?.
In-person community-engaged research protocols should not resume unless standard safety recommendations can be implemented (social distancing, hand washing, mask wearing and, overall, risk mitigation) and contact tracing is in place. For guidance, see the FAQ: What precautions should I take if I am conducting in-person research? Once research meets the phased criteria to resume, travel to and from community sites may be permitted, if compliant with university limitations/restrictions, interaction with members of the public is minimized, and the above safety recommendations are maintained.
What are some issues I should be aware of when resuming community-based research?
Community-engaged research protocols offer special challenges to the health and safety of research personnel and participants. While investigators must follow the same research resumption protocols as their non-community-engaged researcher colleagues (Department Chair/Center Director permission), guidance below is meant to assist generally to promote safety and well-being. For protocol-specific inquiries, contact ORI with the applicable protocol number and request an IRB consult to discuss resumption of research activities.
Investigators must determine appropriate local and community restrictions, requirements, and limitations pertinent to the conduct of the community-engaged research protocol. In an effort to prevent exposure of both potential participants and research teams to the COVID-19 virus, please follow any restrictions or requirements issued for/by the locations/facilities in which the human subjects research takes place.
If the research involves special circumstances or populations (e.g. minors in a school or other settings such as a community center), it is important to abide by the rules of the location but also make sure that parents and others who give consent are fully informed about risks and any measures taken to minimize those risks.
Unlike on-site research interactions, community-based research interactions may reach extensive networks of people, increasing the potential for rapid and mass exposure. Such exposure may make contact tracing with suspected or confirmed cases more challenging.
In the absence of widespread testing, research staff and participants may not know they are COVID-19 positive and could be carriers of the virus. For guidance on pre-screening potential research participants prior to in-person interaction, see the FAQ What precautions should I take if I am conducting in-person research?
When resuming community-engaged research activities, investigators and their teams are encouraged to provide educational resources to their participants. See the UK Community Resources for COVID-19 website.
What precautions should I take if I am conducting in-person research?
As in-person research interactions are permitted under the phases, researchers must consider how to modify in-person participant interactions to reduce the risk of COVID-19 transmission for both participants and the research team.
Researchers must:
- Develop robust testing protocols for staff testing, contact tracing, and self-isolation should a study team member test positive (along with reporting to the Kentucky Department of Public Health).
- Before coming to work, all faculty and staff are required to self-screen each day for signs of COVID-19 symptoms.
- Research staff who work at any UK Healthcare facility complete the UK Healthcare Staff Screening - https://covid-19.ukhc.org/employee-screening-for-covid-19/.
- Employees scheduled to work on-site but who are not feeling well and/or are experiencing any symptoms of illness must stay at home, and immediately contact their supervisor.
- Temperature checks may be performed prior to entering select research spaces or at discretion of investigator.
- Avoid all participants with confirmed or presumptive positive COVID-19 infection or who require potentially aerosolizing procedures, unless this is the focus of the study.
- Require the use of and supply Personal Protective Equipment (PPE). Address training status for nonclinical research personnel who need to be within 6 feet of participants, if appropriate for the study. Address sources of PPE for research use for staff and participants. Address methods to reduce PPE usage by all personnel, including combining research with clinical blood draws.
- Decrease potential exposure for participants and study staff at increased risk of severe illness from COVID-19. People at higher risk include people who:
- are over 60 years of age,
- have underlying health conditions including heart disease, lung disease, or diabetes,
- have weakened immune systems,
- are pregnant,
- are caregivers of children with underlying health conditions or adults who meet any of the criteria listed above.
Inform participants who may be vulnerable to contracting COVID-19 to review the
CDC guidelines for Groups at Higher Risk for Severe Illness. Enable participants to make an informed decision about personal risks related to COVID-19 and respect their wishes.
- Reduce face-to-face interactions with research participants. Strategies may include physical barriers between research personnel and participants, or using technology (e.g., telephone, FaceTime, email, intercoms) to conduct interviews and obtain data.
- Continue remote telework for researchers or research staff whose responsibilities do not require on-site activities (e.g., regulatory specialist, administrative staff, survey researchers). Limit in-person research team meetings to those which allow personnel to maintain physical distancing procedures.
- Develop contingency plans in the event of staff illness or exposure.
- Close access to common areas where possible (e.g., waiting areas, staff break areas, etc.).
- Have a protocol for noncontact screening of participants and study staff for active symptoms of acute respiratory infection possibly related to COVID-19 or high risk of infection. This may be accomplished by communication with participants, care providers, chart review, screening surveys, or other options prior to approaching potential research participants. Individuals with active symptoms or in social isolation (e.g., due to travel history or exposure to someone with COVID-19 symptoms) should be avoided.
- Comply with UK and CDC guidance regarding sanitation of facilities and workspaces.
- Maintain targeted occupancy limits that permit physical distancing. This may require both physical separation of study personnel and staggered schedules, and may require flexibility in work scheduling.
- Stagger participant visits so participants do not congregate at the research site and maintain at least six feet of separation between participants.
Researchers should also:
- Follow any guidelines or instructions from the specific facility (e.g. college, hospital, clinic) where the participant interaction would occur (e.g. screening).
- Consult with the study sponsor, when appropriate.
- Encourage remote monitoring by industry monitors or representatives. In person sponsor visits may not occur until restrictions are lifted by local or institutional authorities.
Should I implement any special procedures in my research space to protect study personnel and research participants from exposure?
Please refer to the VPR Resumption of Research Phased Plan on personnel safety.
Identify the specific sources of COVID-19-related risks. Specific issues to consider include:
- Minimizing the number close interaction or physical contact of participants with your study team or with other participants (e.g., a blood draw or physical exam). The feasibility of physical distancing and maintaining target occupancy limits.
- Use of physical barriers to reduce transmission risks.
- The availability of appropriate personal protective equipment (PPE). Screening before participant interactions.
- Equipment or other items that will be used with multiple participants (e.g., a virtual reality headset, a computer-administered questionnaire, or a sensing device).
- What surfaces will be touched by participants, including items at the research location (e.g., chairs, desktops, research equipment and devices, doorknobs).
- The availability and frequency of disinfection and cleaning procedures for frequently touched surfaces, and adequate hand washing and sanitizing stations, etc.
- If the research procedures include testing participants for SARS-CoV-2 or serological antibodies, identify any restrictions (e.g., CLIA lab analysis) or requirements that apply (e.g., care referral and reporting for positive cases).
- All labs receiving COVID-19 patient specimens must have approval from the Institutional Biosafety Committee. Labs with current approvals for human source material will need to submit an amendment to their protocol. Registration information is available at https://ehs.uky.edu/Biosafety/ and protocols are submitted via https://topaz.uky.edu/. Email biosafety@uky.edu with questions regarding new protocol submissions or amendment creation.
Do I have to call to pre-screen all participants before they come in for a scheduled research visit?
Yes, for continued in-person visits, you must pre-screen all participants prior to the visit. You must call participants prior to their visit to screen them before they arrive and cancel visits for those who give positive responses during the phone screening. Participants must be screened again when they arrive on site.
COVID-19 pre-screening calls do not require submission of a Modification Request as long as done for safety purposes and not research data collection. More details about screening are provided below.
What screening protocol should I use when interacting with research participants?
All research teams are advised to review and comply with facility, location, and/or department procedures for screening all participants.
Research teams are encouraged to initiate these screening procedures in advance of scheduled research visits and to cancel or reschedule visits for persons with current symptoms of a lower respiratory infection. Participants must “pass” screening to be seen in an academic or medical space.
In general, the following screening questions should be asked in advance of the scheduled visit:
- Has the participant had close contact with a confirmed COVID-19 patient within the past 14 days? Close contact is defined by the CDC as:
- being within approximately 6 feet of a COVID-19 case for a prolonged period. Close contact can occur while caring for, living with, visiting, or sharing a health care waiting area or room with a COVID-19 case, or
- having direct contact with infectious secretions of a COVID-19 case (e.g., being coughed or sneezed on).
- Has the participant traveled to any locations confirmed by the Centers for Disease Control and Prevention to have widespread community transmission of COVID-19 in the past 30 days? See https://www.cdc.gov/coronavirus/2019-ncov/travelers/index.html for an updated list of areas with widespread community transmission.
- Does the participant have symptoms of a lower respiratory tract infection (e.g., cough, fever, sore throat, or shortness of breath, or loss of smell/taste) or gastrointestinal symptoms (e.g. diarrhea, nausea)?
If the answer to any of the questions above is “Yes,” terminate the visit and refer the participant to a medical facility for screening and care.
If interaction is taking place in a medical facility, follow medical facility directions on management of COVID-19 positive participants.
Screening outcomes:
- Document participant responses. If all is well, proceed with scheduling the visit and inform participant and/or caregiver regarding applicable visit procedures for on-site screening (e.g., registration screening; temperature checks), requirement to wear masks, and prohibition on bringing guests to the visit.
- If at least two weeks have passed since leaving an area of widespread transmission or exposure to a known COVID-19 case, and they have no symptoms of a lower respiratory tract infection, then the study visit may occur.
- If it is less than two weeks since their potential COVID-19 exposure and the participant has no symptoms, the study investigator should assess the potential risks and benefits of continuing the research visit versus rescheduling the visit until after the two-week period, if feasible.
- If the participant has symptoms of a lower respiratory tract infection, the investigator should consult the appropriate hospital resources to determine the next steps for evaluation and management.
Once participant is on-site, conduct screening procedures consistent with facility requirements, unless screening performed by facility staff upon entry or registration staff.
Are positive COVID-19 test results reportable to the state of Kentucky? (Updated 10/14/20)
Yes, positive or presumptive positive SARS-CoV-2 results must be reported to public health authorities in the state of Kentucky. They may not accept results that come from a lab which is not certified by the federal CLIA program or state equivalent. If you obtain a positive result, report it to the state of Kentucky and, if required, UK HealthCare. If the test is not a recognized/verified test, you should tell the participant about the result and the limits of that test, then encourage them to get a valid test from a healthcare facility. Find instructions under the “Healthcare Providers” section under “Resources” on the Kentucky Public Health COVID-19 website: https://govstatus.egov.com/kycovid19.
Guidance from the Office for Human Research Protections (OHRP) Guidance on COVID-19 states: “Legally Required Reporting: When required by law to provide information related to an individual's COVID-19 test results to a public health authority, including individually identifiable information about individuals who are research participants, the HHS protection of human subjects regulations do not prevent investigators or institutions from fulfilling this requirement (even if doing so would be inconsistent with statements made in the study's consent form). The existence of a Certificate of Confidentiality[3] does not alter an investigator's ability to disclose a research participant's COVID-19 test results when required by federal, state, or local laws. For example, if a research participant tests positive for COVID-19, an investigator may provide this test result to a public health authority if required to do so under applicable state or federal law. In such circumstances, investigators should inform the participant of the required reporting of results.” [https://www.hhs.gov/ohrp/regulations-and-policy/guidance/ohrp-guidance-on-covid-19/index.html]
For Questions About Screening, Testing or Tracing? Contact Our Health Corps.
Call 859-218-SAFE(7233)
Email healthcorps@uky.edu
Hours of Operations:
Monday-Friday 8 a.m. - 8 p.m.
Saturday-Sunday 10 a.m. - 5 p.m.
After research reopens, if a subject or research team member is discovered to have tested COVID positive or presumptive positive with 2 weeks of an in-person research visit, would we promptly report to the IRB as an Unanticipated Problem involving Risks to Subjects or Others (UPIRSO)?
An Unanticipated problem involving risks to subjects or others (UPIRSO)- includes any incident, experience, or outcome that meets all of the following criteria:
- Unexpected (in terms of nature, severity, or frequency) given (a) the research procedures that are described in the protocol-related documents, such as the IRB-approved research protocol and informed consent document; and (b) the characteristics of the subject population being studied;
Consider whether unexpected in frequency given the presumed measures that in place to mitigate risks. Might be expected for COVID-19 research. - Related or possibly related to participation in the research; and
Could it have occurred because of an in-person research interaction/intervention? Likely if interaction is for research and not concurrent with other interaction. - Suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized.
Likely so, as suggests others at greater risk of virus transmission than was previously known or recognized.
What do I need to know to submit/conduct COVID-19 research?
The document below includes information on submitting new or updating existing research which focuses on COVID-19.
Conduct and Submission of COVID-19 Research in Human Subjects [PDF]
What is the PREP Act and what should subjects in COVID-19 interventional trials be told in the informed consent about liability limits?
On March 10, 2020, the HHS Secretary issued a Public Readiness and Emergency Preparedness (PREP) Act Declaration which was amended effective March 27, 2020 that provides liability immunity (except for willful misconduct) for claims of loss caused, arising out of, relating to, or resulting from manufacture, distribution, administration or use of countermeasures to diseases, threats, and conditions determined by the Secretary to constitute a public health emergency to entities and individuals involved in the development, manufacture, testing, distribution, administration, and use of such countermeasures.
PREP limits legal rights to sue covered persons engaged in select COVID-19 countermeasures (including select treatment research) and provides compensation for eligible individuals who suffer injuries from use of covered products.
Covered Countermeasures may include vaccines, drugs, or medical devices to be used to treat, diagnose, cure, prevent, or mitigate COVID-19.
Products must be:
- a qualified pandemic or epidemic product;
- a security countermeasure;
- a respiratory protective device approved by NIOSH;
- approved, licensed, or cleared by FDA;
- authorized under an Emergency Use Authorization (EUA) issued by FDA;
- described in an Emergency Use Instructions (EUI) issued by the CDC; or
- used under either an Investigational New Drug (IND) application or an Investigational Device Exemption (IDE).
In addition to meeting the scope of the act, reasonable precautions must be taken to facilitate the safe use of covered countermeasures. See the PREP Act Q&As and PREP Act Glossary for detailed information.
CONSENT LANGUAGE:
The following statement should be added to the informed consent for applicable COVID-19 studies, in order to inform participants regarding their legal rights.
Due to the coronavirus public health emergency, the federal government has issued an order that may limit your right to sue if you are injured or harmed while participating in this COVID-19 study. If the order applies, it limits your right to sue and recover losses from the researchers, healthcare providers, any study sponsor, distributor or manufacturer involved with the study. However, the federal government has a program that may provide compensation to you or your family if you experience serious physical injuries or death. To find out more about this “Countermeasures Injury Compensation Program” please go to https://www.hrsa.gov/cicp/about/index.html or call 1-855-266-2427.
If participants have questions about the Countermeasures Injury Compensation program (CICP) direct them to the CICP website which provides Requester FAQs, Fact Sheet and contact information.
Are there other resources I can view for further guidance? (Updated 1/15/21)
- UK Vice President for Research (VPR) COVID-19 Guidance for Researchers - https://www.research.uky.edu/resources/covid-19-guidance-researchers
- UK Institutional Biosafety COVID-19 Guidance - https://ehs.uky.edu/biosafety/ (Scroll down to COVID-19)
- UK Healthcare COVID-19 Website - https://covid-19.ukhc.org/ includes guidelines, TeleCare Toolkit, FAQs, Infection Prevention and Control, UKHC Employee Screening and Resources.
- UK Healthcare Staff Screening - https://covid-19.ukhc.org/employee-screening-for-covid-19/
- UK COVID-19 FAQs - https://www.uky.edu/coronavirus/faqs
- Employee and Human Resources Information - https://www.uky.edu/coronavirus/faqs#hr
- Campus Restart - https://www.uky.edu/coronavirus/faqs#restart
- Kentucky Public Health - https://govstatus.egov.com/kycovid19 (Scroll to RESOURCES for Guidance by Topic and Healthcare Provider Information on Case Reporting)
- Kentucky COVID-19 hotline provides guidance for individuals in the state of Kentucky with symptoms 800-722-5725
- Centers for Disease Control and Prevention Coronavirus website https://www.cdc.gov/coronavirus/2019-ncov/index.html
- Council on Governmental Relations (COGR) Research Ethics & Compliance Committee - Human Subjects FAQs May 4, 2020 (Version 1.0)
https://www.cogr.edu/sites/default/files/FAQS%20ON%20HUMAN%20SUBJECTS%20for%20REC%20final%20may%204%202020.pdf - Food and Drug Administration (FDA) "Assessing COVID-19-Related Symptoms in Outpatient Adult and Adolescent Subjects in Clinical Trials of Drugs and Biological Products for COVID-19 Prevention or Treatment" https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-covid-19-related-symptoms-outpatient-adult-and-adolescent-subjects-clinical-trials-drugs
- "FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency" (Updated September 21, 2020)https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-guidance-conduct-clinical-trials-medical-products-during-covid-19-public-health-emergency
- Food and Drug Administration (FDA) "FDA Digital Health Policies and Public Health Solutions for COVID-19"
https://www.fda.gov/medical-devices/digital-health/digital-health-policies-and-public-health-solutions-covid-19 - National Cancer Institute Central IRB (NCI-CIRB) "Memorandum On Interim Guidance For Patients On Clinical Trials Supported By The NCI Cancer Therapy Evaluation Program And The NCI Community Oncology Research Program (NCORP)"
https://www.ncicirb.org/announcements/memorandum-interim-guidance-patients-clinical-trials-supported-nci-cancer-therapy - National Cancer Institute Central IRB (NCI-CIRB) "Interim Guidance for Patients on Clinical Trials Supported by the NCI Cancer Therapy Evaluation Program and the NCI Community Oncology Research Program (NCORP)" (March 13, 2020)
https://www.ncicirb.org/announcements/memorandum-interim-guidance-patients-clinical-trials-supported-nci-cancer-therapy - National Cancer Institute Central IRB (NCI-CIRB) "Additional Guidance Regarding Alternative Procedures for Clinical Trials Supported by the NCI Cancer Therapy Evaluation Program(CTEP) and NCI Community Oncology Research Program (NCORP) Affected by the Spread of the Novel Coronavirus" (March 23, 2020)
https://ncicirb.org/sites/ncicirb/files/Memo_Additional_Guidance_Clinical_Trial_Activities_Affected_Coronavirus.pdf - National Institutes of Health (NIH) has issued notices on the NIH Extramural Response to Natural Disasters and Other Emergencies website
https://grants.nih.gov/grants/natural_disasters.htm - Office for Civil Rights (OCR) "Notification of Enforcement Discretion for Telehealth Remote Communications During the COVID-19 Nationwide Public Health Emergency"
https://www.hhs.gov/hipaa/for-professionals/special-topics/emergency-preparedness/notification-enforcement-discretion-telehealth/index.html - Office for Human Research Protections (OHRP) Guidance on COVID-19 - https://www.hhs.gov/ohrp/regulations-and-policy/guidance/ohrp-guidance-on-covid-19/index.html
What if my research involves a public health authority or public health surveillance?
If you think your work is public health surveillance in addition to (or instead of) research, please fill out a UK Not Human Subjects Research (NHR) form after obtaining documentation (e.g., email) that describes the specifics of any involvement of a public health authority. You should include the following in the summary of the NHR form:
- a brief description of the role of the public health authority (e.g., design, coordination, direction);
- clarification of whether the public health authority plans to use the data to make decisions; and
- if so, when and for what purpose the data will be used.
This information is important because it impacts the type of IRB review required (if any).
What public health surveillance activities are deemed not to be human subject research under the revised Common Rule?
- The activity must be a public health surveillance activity (45 CFR 46.102(l)(2));
- The activity must be conducted, supported, requested, ordered, required, or authorized by a public health authority (45 CFR 46.102(k) and 46.102(l)(2)); and
- The activity must be limited to that necessary to allow a public health authority to identify, monitor, assess, or investigate potential public health signals, onsets of disease outbreaks, or conditions of public health importance (including trends, signals, risk factors, patterns in diseases, or increases in injuries from using consumer products) (45 CFR 46.102(l)(2)).
However, FDA regulations may apply if this involves use of an investigational in-vitro diagnostic device.
Detailed guidance is available from the Office for Human Research Protections (OHRP).
Can I submit a new research project? (Added 3/26/20)
Yes. The IRB will continue to review new study submissions through E-IRB. Priority will be given to research that involves COVID-19.
Depending on the study procedures, you may be asked to replace or delay use of in-person data collection consistent with institutional directives to reduce exposure to COVID-19.
Should I indicate if my research project is related to COVID-19 in my project title? (Added 4/16/20)
***Effective April 16, 2020: If your research involves investigating any aspect of COVID-19, please enter “COVID-19” at the start of your Project Title in your IRB protocol application.***
Please add COVID-19 to “Title of Project” and “Short Title Description” of your current IRB protocol.
For protocols being reviewed by an external IRB, please enter “RELIANCE COVID-19” before the PROJECT and SHORT titles.
Is there a biobank for UK Researchers specifically for research on COVID-19? (Added 4/23/20)
Yes, an announcement from CCTS on April 23, 2020 states:
In response to the COVID-19 pandemic, the University of Kentucky Center for Clinical and Translational Science (CCTS) has created a new biospecimen bank to support research on the novel virus. Logistical support for the biobank is led by the UK Markey Cancer Center.
The COVID-19 Research Registry and Specimen Bank will collect specimens from volunteers who have tested positive for or are suspected of having the virus. The bank will work closely with the UK CURE (COVID-19 United Research Experts) Alliance, a new workgroup within UK’s College of Medicine that unites experts from across campus to focus on advising COVID-19 patient care and clinical trials.
Samples can be requested through the CCTS Service Request Form by selecting "Biospecimens" and then "COVID-19 Biobank."
Can I still enroll patients in clinical trials?
Enrollment in clinical trials should be placed on hold immediately, except where treatment during a clinical trial is medically necessary or of potential benefit for the patient, such as some oncology studies and device studies and some clinical trials with investigational drugs. Trials with drugs or devices should continue to enroll new subjects only if there are no other treatments available and the subjects would be placed at risk without treatment. Clinical trials with placebo arms should be placed on enrollment hold.
If suspending new enrollment, contact industry sponsors of these trials immediately and inform them that enrollment for their study is on hold or will experience limited enrollment for up to 90 days if applicable. Document this communication with the industry sponsors.
Can studies being conducted entirely during an inpatient admission continue?
Inpatient studies that do not involve direct patient benefit, or that are not part of COVID-19 research efforts, should be paused. Clinical care of our patients with COVID-19 and other conditions is now the priority. The goal here is to free up all staff to respond to this and to minimize patient contact by non-essential personnel when possible. Examples of inpatient studies that should be paused include:
- Comparative effectiveness studies randomizing patients to a placebo/non-intervention arm versus alternative clinical management;
- Implementation science trials looking at changes in clinical management;
- Survey research on patient perceptions.
Is an IRB Modification Request required to delay, pause, or temporarily modify research activities due to COVID-19? (Added 4/10/20)
Research protocols or any single component of a research protocol may be delayed/paused or temporarily modified for COVID-19 concerns without submitting a Modification Request to the IRB. Per federal regulations, an investigator can make changes to a protocol to eliminate apparent hazards to research participants without first obtaining IRB approval. The change may apply to a single participant or all participants enrolled in the research study.
However, if changes to research procedures are permanent or propose new COVID research and not currently approved by the IRB, a Modification Request should be submitted prior to implementing the changes.
For FDA studies, please see next question.
Does FDA require sites to track and report changes that were made without IRB approval to minimize hazards from COVID-19? (Added 3/26/20)
Yes. Changes to protect the life and well-being of research subjects (e.g., to limit exposure to COVID-19) may be made without IRB approval, but FDA-regulated studies require such changes to be reported afterwards. Investigators should submit an aggregate account of changes implemented without prior IRB approval on one protocol violation report.
FDA has advised sponsors to collect a list of contingency measures taken; list subjects affected by study number; and indicate the impact on safety or efficacy results, if applicable. Check with the study sponsor on reporting requirements.
Clinical trials that have paused recruitment due to COVID-19, should update their study status on ClinicalTrials.gov record to “active, not recruiting” within 30 days. For questions regarding ClinicalTrials.gov, please contact Emily Bradford at emily.bradford@uky.edu.
Do I need to notify the IRB if all study activities are paused?
Not at this time. You must notify the study sponsor (see below). Please track when you pause all study activities and document all missed visits, procedures, appointments, and other relevant study activities. We are working on a mechanism to report these in aggregate at some point in the near future, rather than reporting each event individually.
Do I need to notify my study subjects of the pause?
Yes. All active study subjects need to be notified of the pause, and what it means for them. We recommend that you do this by phone, but mail and e-mail (if you have consent for email contact) may also be used if needed under these circumstances. You do not need an IRB modification for this notification.
What should I tell study subjects?
The following sample script may be adapted to fit your particular study:
Hello, <subject name>. I am calling you today to let you know that several actions have been put into place regarding research/clinical trials at the University of Kentucky and all of its locations. Some of these may affect you. These include:
- New enrollment in clinical trials will be limited until further notice. If you are screening or have recently consented to participate in a clinical trial, please contact ____________.
- If you are in treatment on a clinical trial, you may continue treatment, although the study visit schedule may change. When you come for your treatment, please be prepared to answer questions about your recent trips and health. This screening is intended to protect you, your family, and our healthcare workers from coronavirus infection. If you are sick, have symptoms, or have had significant exposure to somebody who is infected with coronavirus, your visit may be rescheduled. We will do everything we can to maintain your treatment schedule.
- If you are having follow up visits, we may reschedule or cancel these visits during the next 90 days or until further notice. These are not treatment visits. A research team member will contact you prior to your next visit.
We understand that you may have concerns about your participation in clinical trials. Please contact <study coordinator> for specific concerns. If you are experiencing adverse events, or have been diagnosed with coronavirus infection, please contact your study team immediately using the 24-hour contact number in your consent form.
Document what you have communicated to your study subjects, the purpose for the communication (COVID 19), and when it was communicated.
Do I need to re-consent subjects on changes to protect from COVID-19? (Added 3/26/20)
In most cases, new information regarding changes to protect from COVID-19 may be provided via a letter or other form of communication. Subjects must be provided with significant new findings that develop during the research which may relate to their willingness to continue participation. However, re-consent is not necessary unless the changes to the research are such that the original consent is no longer valid. See the ORI New Information Guidance for help determining whether notification is adequate for providing new information or re-consent is warranted.
Do I need approval from the IRB for communications to study subjects explaining the pause in activities?
No. It is not necessary to submit a modification. Keep documentation of dates, times and subjects who were contacted.
What guidance is available for e-consent or remote consent? (Updated 2/25/21)
Remote informed consent may be proposed for new COVID-19 research or implemented for an existing protocol in response to the pandemic. Electronic consent may be proposed for in-person or remote consent. The ORI Outline of FDA Guidance for Industry: Use of Electronic Informed Consent in Clinical Investigations summarizes federal guidance and consideration of system security, identity verification, legally valid e-signatures, and the provision of subject copies.
For options conducting remote consent for subjects who cannot travel or patients in isolation, see the UK ORI Remote Informed Consent Guidance or questions 10 & 11 in the FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency.
The University of Kentucky Center for Clinical and Translational Science (CCTS) has presented a REDCap mobile app and consent feature webinar. In addition, they have shared a video demo of their process for obtaining remote consent of UK COVID-19 Biobank participants using REDCap.
What if I have additional questions about my study?
For Clinical Research questions: You may contact the Center for Clinical and Translational Sciences by email at CCTS@uky.edu or phone at (859) 323-3775 and your question will be forwarded to a content expert who will try to answer your question.
For General IRB or Human Research questions: You may contact the Office of Research Integrity by email at IRBSubmission@uky.edu or phone at (859) 257-9428 and your question will be forwarded to the appropriate staff member. If your question is about a particular study, please include the IRB protocol number and PI name.
What e-consent options are FDA Part 11 compliant for FDA-regulated clinical trials? (Updated 10/9/20)
Software may not be compliant with FDA Part 11 out of the box. It may need to be configured and processes should be implemented to ensure use is in a manner that meets Part 11 requirements, including identity verification, signature certification, and audit trails. Below is information and resources on potential options. Note: this does not constitute approval or endorsement of the IRB. Also, the customer retains responsibilities for implementation of features, functions, and training in order to meet regulatory compliance, implemented features and processes.
DocuSign
Docusign’s Part 11 module website provides an overview and operational information on administration, settings, and features. DocuSign's Part 11 module contains industry-designed capabilities that include:
- Pre-packaged account configuration
- Signature-level credentialing
- Signature-level Signing Reason
- Signature manifestation (Printed Name, Time Stamp, and Signature Reason)
- Detailed audit trail
- Tamper-evident digital seal
Adobe Sign
Adobe Sign is a cloud-based e-signature service which supports electronic and digital signatures. Consent documents may be uploaded into the Adobe Sign portal and sent by email to prospective participants. For each feature, the Adobe Sign and 21 CFR Part 11 guide, prepared by Montrium Consulting, describes how it complies and the associated shared customer responsibilities.
FDA COVID MyStudies Application (App)
FDA has made previously developed platform to obtain informed consent securely when face-to-face contact is not possible or practical due to COVID-19 control measures. Investigators download COVID MyStudies in the Apple App and Google Play. The app may be used to send informed consent documents to individuals or LARs. Investigators and participant receive copies of completed digital informed consent documents.
Does UK provide general guidance for study subjects, in addition to that provided by individual study staff? (Added 3/26/20)
Yes. The UK Center for Clinical and Translational Science (CCTS) provides general Guidance for Research Participants during COVID-19.
Do I need to notify the industry sponsor of modifying or pausing my clinical trial? (Updated 3/26/20)
Yes. Contact your study sponsor to discuss how best to move forward with performing remote research visits, when possible. Given that we expect this to be a temporary situation, a protocol modification is not necessary. Sponsors may grant protocol waivers to conduct research in this manner and allow for missed assessments and missed visits to be documented and reported later as this is a required change to research to eliminate apparent hazards to research subjects. If the study is paused, the sponsor will need to be notified.
What guidance does FDA provide for clinical trials during the COVID-19 Pandemic? (Updated 9/22/20)
The FDA outlines considerations to assist sponsors in assuring the safety of trial participants, maintaining compliance with good clinical practice and minimizing risks to trial integrity. Considerations recommended include, among others, sponsors evaluating alternative methods for assessments, like phone contacts or virtual visits and offering additional safety monitoring for those trial participants who may no longer have access to investigational product or the investigational site.
For more information visit:
"FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency" (Updated September 21, 2020)
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-guidance-conduct-clinical-trials-medical-products-during-covid-19-public-health-emergency
Other FDA Guidance:
"Assessing COVID-19-Related Symptoms in Outpatient Adult and Adolescent Subjects in Clinical Trials of Drugs and Biological Products for COVID-19 Prevention or Treatment"
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-covid-19-related-symptoms-outpatient-adult-and-adolescent-subjects-clinical-trials-drugs
"FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency"
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-guidance-conduct-clinical-trials-medical-products-during-covid-19-public-health-emergency
Has the NIH issued any formal guidance on clinical trials or other human subjects research activities?
Yes, although at this point in time the guidance is broad and geared predominately at administrative issues.
Guidance can be found on the NIH web page for Natural Disasters:
https://grants.nih.gov/grants/natural_disasters.htm.
What should I do to ensure that subjects in clinical trials remaining open have an adequate supply of study drug? (Updated 5/18/20)
For clinical trials with investigational drugs, devices and biologics, please confirm with the sponsor that the supply is not at risk. Please review investigational supplies and identify any that will expire within the next 90 days and communicate with the sponsor regarding resupply measures.
The UK Investigational Drug Service has standardized procedures for shipping investigational drugs to subjects.
What is the suggested COVID-19 screening protocol for study subject visits?
Call every subject the day before a site or home visit and ask the following travel screening questions:
- Have you or your family members traveled in the last 14 days?
If yes, ask for dates and locations of travel. If travel was outside of the local upstate region you may elect to postpone the visit by two weeks. - Do you or anyone in your home have any of the following symptoms: cough, fever, shortness of breath? Have you or anyone in your family had any contact with a confirmed COVID-19 patient?
If yes to either question, then postpone the in-person visit for at least two weeks and make sure they have spoken with a health care professional. - All subjects should be re-screened when they arrive for their visit.
If a subject presents with the symptoms listed above at their study visit, terminate the visit and have them contact their primary provider.
What should I do about missed visits for trials that remain open?
If a subject misses a scheduled visit during this period, it should be documented including the reason for the missed visit (ex. COVID-19). Contact the study sponsor for guidance on how they would like such notifications and documentation handled. It is likely, given that this will be common during this national emergency, that notification requirements will be given to all study sites.
What should I do to prepare for study subjects being removed from active protocols due to COVID-19 infection?
Review each clinical trial protocol for drug reductions or holds due to serious adverse events or for other reasons. Subject who are diagnosed with COVID-19 will likely be removed from study treatment by the sponsor or investigator. Treatment schedule modifications or removal from the study for subjects who are in quarantine should be discussed with the sponsor. Items should be reported consistent with the UK IRB reporting policies.
Should I modify my study protocol to reduce risk to my study subjects?
When possible, you should consider such modifications.
Eliminating immediate risks may include actions to reduce potential exposure to COVID-19 or to continue to provide medically necessary study care (including study drug) to subjects who have been placed in isolation or quarantine because of suspected or known exposures. We encourage investigators to take necessary steps to eliminate additional risks to subjects. Even in studies that offer benefit to the patient, investigators should balance the potential benefits with the risk of patient exposure to COVID-19 by further participation.
Depending on the protocol, exposure or a positive test for COVID-19 could be classified as an adverse event, and could also cause a protocol-required reduction or temporary or permanent discontinuation of investigational drug. Giving study drug to isolated or quarantined subjects will also be situational. For example, IV infusions could pose a serious problem if given outside the clinic. The protocols will often indicate under which conditions drugs should be discontinued and how. Death during a study is a serious adverse event.
We recommend that study teams communicate with sponsors/clinical research organizations about these scenarios and document the responses.
What if the sponsor issues new guidance or information about COVID-19 risks for my study?
You will need to submit a modification request to the IRB. Your study may be paused if the sponsor, Data Safety Monitoring Board (DSMB) or you identify any new increased risk to study subjects related to the protocol.
What should I do about study/sponsor monitor visits? (Updated 8/26/20)
To the extent possible, all monitoring visits, site initiation visits, site qualification visits, should occur remotely.
Otherwise, communicate facility safety requirements to the study sponsor and/or clinical research organization, incorporate barriers or social distancing when making site-visit arrangements, and perform safety screening of on-site monitors prior to visits.
Document the contact in the study documentation.
What facilities exist for virtual visits with study subjects or study monitors? (Updated 8/26/20)
Depending on the recipients' resources, communications options include telephone conferencing, UK Zoom, UK Telehealth, REDCap, and other survey platforms.
The Center for Clinical and Translational Science (CCTS) provides a procedure for obtaining view-only access to the Electronic Medical Record (EMR) and offers options for monitoring documents outside of the EMR using existing platforms at UK (One Drive and Zoom). EMR access is enabled by UK HealthCare IT and is subject to review and/or audit by the Office of Corporate Compliance. Details and resources for remote monitoring are available at ccts.uky.edu/RemoteMonitoring.
What should I do about study samples that need to be shipped to the sponsor?
We recommend contacting the sponsor to delay the shipment, unless the samples cannot be stored.
Will the IRB continue to accept and review Other Events, Annual Administrative Review, and Continuing Review submissions? (Added 3/26/20)
Yes. The IRB will continue to review these submissions. AARs and CRs will be approved with the expectation that ongoing research is compliant with applicable restrictions and accommodations for reducing exposure to COVID-19.
Do I have to submit Annual Administrative Review and Continuing Review submissions during pause? (Added 3/26/20)
Yes, as long as you are able to submit the AAR or CR without endangering yourself or staff, the IRB expects to receive these reviews. If you expect delays, notify the Office of Research Integrity at 859-257-9428 or IRBSubmission@uky.edu.
Are ORI Quality Assurance Wellness Checks being conducted at this time? (Added 4/7/20)
The ORI Quality Improvement/Quality Assurance (QA/QI) Program is not scheduling or conducting any visits at this time due to COVID-19 concerns. We will resume our visits once the university returns to regular business function and the conduct of in-person events.
Investigators who had Wellness Check visits scheduled for late March and all of April 2020 have been contacted and informed their visit will be delayed until after the current COVID-19 restrictions have been lifted and staff/students return to campus. We will work with investigators to reschedule their visit as soon as possible.
Please contact Pam Stafford pastaf3@uky.edu or Kasandra Lambert kasandra.lambert@uky.edu if you have questions or concerns.
Are IRB meetings being conducted remotely? (Added 4/7/20)
Yes. Due to current COVID-19 concerns, IRB meetings are being conducted electronically through Zoom. Investigators will receive an invite containing a Zoom link prior to the meeting.
Investigators should inform ORI staff if they have other study personnel to whom they need a meeting invitation sent. Investigators should not forward the meeting link to their study personnel. It’s important for ORI staff to know everyone who will be attending so they are aware of who is and is not supposed to be admitted into the virtual meeting during each scheduled appointment time.
In addition, the number of attendees for each protocol needs to be limited in order to keep attendance tracking manageable for documentation purposes, so we ask that investigators have no more than two (2) individuals from the list of study personnel join them for the meeting.
What if my protocol was reviewed by an external IRB? (Added 3/26/20)
Check with reviewing IRB on reporting requirements as they may differ with local IRB
Page created 3/19/2020 10:00 am
Updated 6/11/2021 1:45 pm